

Multiple myeloma (MM) is defined by the clonal expansion of malignant plasma cells in the bone marrow (BM) and has a 5-year survival rate of 50% (Siegel el al. 2018, Cancer J. Clin.). MM remains incurable due to the development of resistance to current chemotherapies; therefore, it is paramount to investigate novel treatments and the mechanisms of drug resistance in MM cells. Interestingly, obesity correlates with increased incidence of MM and high body mass index correlates with a poor treatment response (Marinac et al. 2019, JNCI Cancer Spectr, Groß et al. 2017, Oncotarget). Obesity is a major risk factor for many cancers, however, given the complexity of obesity, there are an array of mechanisms by which obesity may support tumor cells. Studies of obesity and MM are mainly at the epidemiological level and have not extensively explored the mechanism of this relationship. Therefore, there is a critical need to understand how obesity contributes to support cancers such as MM. Possible mechanisms may be through the increased availability of free fatty acids or through other factors that are found in obese patients. We hypothesize that lipid metabolism contributes to obesity-linked cancers such as MM.
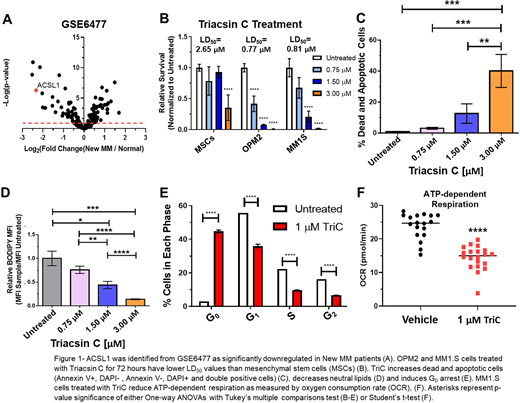
Recently, changes in lipid metabolism have been shown to support the proliferation, migration and the development of drug resistance in other blood cancers such as acute myeloid leukemia (Tabe et al. 2017, Cancer Res, Tabe et al. 2018, Sci. Reports) and solid tumors such as breast (Wang et al. 2017, JCI Insights) and prostate (Mitra et al. 2017, BMC Cancer) cancer. However, the role of lipid metabolism in MM cells has been understudied. Therefore, we hypothesized that genes within the Hallmark Fatty Acid Metabolism gene set (https://www.gsea-msigdb.org) would be differentially expressed between healthy patients and those with MM. We mined the clinical data (GSE6477, Chng et al. 2007, Cancer Res.) and found that transcripts of an enzyme critical for lipid metabolism, acyl-CoA synthetase long chain member 1 (ACSL1), was significantly downregulated (Figure 1A, Log2(Fold Change)=-2.33, adjusted p value=1.64*10-5, false discovery rate) in patients with newly diagnosed MM relative to normal plasma cells. Therefore, we hypothesized that ACSL1 may act as a tumor suppressor in MM.
In order to test the role of the ACSL family as tumor suppressors, we treated human (MM1.S, OPM2 and RPMI-8226) and mouse myeloma (5TGM1) cell lines with an inhibitor (Triacsin C, TriC) of four of the five human acyl-CoA synthetase long chain family members (ACSL1,3,4 and 5). Contrary to our hypothesis, TriC treatment significantly decreased MM cell proliferation (Figure 1B, p<0.0001, One-way ANOVA Tukey's multiple comparisons test is used throughout unless otherwise noted), increased apoptosis (Figure 1C, p<0.001) and caused G0 arrest (Figure1D, p<0.0001) in a dose-dependent manner. Motivated to understand if TriC's toxicity was due to changes in metabolic dynamics, MM1.S cells were treated with 1 μM TriC for 30 minutes and subjected to a metabolic flux assay (Seahorse XF, Agilent). TriC treatment significantly reduced ATP-dependent respiration from fatty acid oxidation (FAO) (Figure 1F, p<0.0001 Student's t-test) and increased proton leak (p<0.0001).
Taken together, our data demonstrate that TriC-mediated ACSL inhibition in MM cells decreases proliferation, induces G0 arrest, apoptosis and decreases FAO-dependent respiration and mitochondrial function. It is unclear what ACSL family member is responsible for the phenotype we report here. To address these questions, future studies will focus on genetically targeting individual ACSL family members and characterizing the lipidomic profile of MM ACSL mutants. Our data also suggests that fatty acids are used as an energy source, therefore we will explore how FAO contributes to MM cell proliferation and survival.
No relevant conflicts of interest to declare.

